

Introduction¶
LaminDB is an open-source data framework for biology.
Manage storage & databases with a unified Python API (“lakehouse”).
Track data lineage across notebooks & pipelines.
Integrate registries for experimental metadata & in-house ontologies.
Validate, standardize & annotate.
Collaborate across distributed databases.
LaminDB features
Actual content in lamin-docs.
LaminHub is a data collaboration hub built on LaminDB similar to how GitHub is built on git.
LaminHub features
Actual content in lamin-docs.
Basic features of LaminHub are free. Enterprise features hosted in your or our infrastructure are available on a paid plan!
Quickstart¶
You’ll ingest a small dataset while tracking data lineage, and learn how to validate, annotate, query & search.
Setup¶
Install the lamindb Python package:
pip install 'lamindb[jupyter,bionty]'
Initialize a LaminDB instance mounting plugin bionty for biological types.
# store artifacts in a local directory `./lamin-intro`
!lamin init --storage ./lamin-intro --schema bionty
Show code cell output
❗ using anonymous user (to identify, call: lamin login)
💡 connected lamindb: anonymous/lamin-intro
Track¶
Run track() to track the inputs and outputs of your code (Transform).
When you first run ln.track(), it creates a stem_uid & version to unambiguously identify your notebook or script.
import lamindb as ln
# copy-pasted identifiers for your notebook or script
ln.settings.transform.stem_uid = "FPnfDtJz8qbE" # <-- auto-generated by running ln.track()
ln.settings.transform.version = "1" # <-- auto-generated by running ln.track()
# track the execution of your notebook or script
ln.track()
Show code cell output
💡 connected lamindb: anonymous/lamin-intro
💡 notebook imports: anndata==0.10.7 bionty==0.43.1 lamindb==0.72.1 pandas==1.5.3
💡 saved: Transform(uid='FPnfDtJz8qbE5zKv', version='1', name='Introduction', key='introduction', type='notebook', created_by_id=1, updated_at='2024-05-25 15:24:51 UTC')
💡 saved: Run(uid='kU0ewjMF8cXWvemZBo5f', transform_id=1, created_by_id=1)
Run(uid='kU0ewjMF8cXWvemZBo5f', started_at='2024-05-25 15:24:51 UTC', is_consecutive=True, transform_id=1, created_by_id=1)
Artifacts¶
Use Artifact to manage data in local or remote storage.
import pandas as pd
# a sample dataset
df = pd.DataFrame(
{"CD8A": [1, 2, 3], "CD4": [3, 4, 5], "CD14": [5, 6, 7], "perturbation": ["DMSO", "IFNG", "DMSO"]},
index=["observation1", "observation2", "observation3"],
)
# create an artifact from a DataFrame
artifact = ln.Artifact.from_df(df, description="my RNA-seq", version="1")
# artifacts come with typed, relational metadata
artifact.describe()
# save data & metadata in one operation
artifact.save()
Show code cell output
Artifact(uid='NtOFwmJMkuNYO5rjoQUS', version='1', description='my RNA-seq', suffix='.parquet', accessor='DataFrame', size=4122, hash='EzUJIW3AamdtaNxG_Bu_nA', hash_type='md5', visibility=1, key_is_virtual=True)
Provenance
.created_by = 'anonymous'
.storage = '/home/runner/work/lamindb/lamindb/docs/lamin-intro'
.transform = 'Introduction'
.run = '2024-05-25 15:24:51 UTC'
Artifact(uid='NtOFwmJMkuNYO5rjoQUS', version='1', description='my RNA-seq', suffix='.parquet', accessor='DataFrame', size=4122, hash='EzUJIW3AamdtaNxG_Bu_nA', hash_type='md5', visibility=1, key_is_virtual=True, created_by_id=1, storage_id=1, transform_id=1, run_id=1, updated_at='2024-05-25 15:24:52 UTC')
View data lineage:
artifact.view_lineage()
Show code cell output

Load an artifact:
artifact.load()
Show code cell output
| CD8A | CD4 | CD14 | perturbation | |
|---|---|---|---|---|
| observation1 | 1 | 3 | 5 | DMSO |
| observation2 | 2 | 4 | 6 | IFNG |
| observation3 | 3 | 5 | 7 | DMSO |
An artifact stores a dataset or model as either a file or a folder.
How do I register a file or folder?
Local:
ln.Artifact("./my_data.fcs", description="my flow cytometry file")
ln.Artifact("./my_images/", description="my folder of images")
Remote:
ln.Artifact("s3://my-bucket/my_data.fcs", description="my flow cytometry file")
ln.Artifact("s3://my-bucket/my_images/", description="my folder of images")
You can also use other remote file systems supported by fsspec.
How does LaminDB compare to a file system or object store?
Similar to organizing files in file systems & object stores with paths, you can organize artifacts using the key parameter of Artifact.
However, LaminDB encourages you to not rely on semantic keys but instead organize your data based on metadata.
Rather than memorizing names of folders and files, you find data via the entities you care about: people, code, experiments, genes, proteins, cell types, etc.
LaminDB embeds each artifact into rich relational metadata and indexes them in storage with a universal ID (uid).
This scales much better than semantic keys, which lead to deep hierarchical information structures that can become hard to navigate.
Are artifacts aware of array-like data?
Yes.
You can make artifacts from paths referencing array-like objects:
ln.Artifact("./my_anndata.h5ad", description="annotated array")
ln.Artifact("./my_zarr_array/", description="my zarr array store")
Or from in-memory objects:
ln.Artifact.from_df(df, description="my dataframe")
ln.Artifact.from_anndata(adata, description="annotated array")
How to version artifacts?
Every artifact is auto-versioned by its hash.
You can also pass a human-readable version field and make new versions via:
artifact_v2 = ln.Artifact("my_path", is_new_version_of=artifact_v1)
Artifacts of the same version family share the same stem uid (the first 16 characters of the uid).
You can see all versions of an artifact via artifact.versions.
Labels¶
Label an artifact with a ULabel.
# create & save a label
candidate_marker_study = ln.ULabel(name="Candidate marker study").save()
# label an artifact
artifact.labels.add(candidate_marker_study)
artifact.describe()
# the ULabel registry
ln.ULabel.df()
Show code cell output
Artifact(uid='NtOFwmJMkuNYO5rjoQUS', version='1', description='my RNA-seq', suffix='.parquet', accessor='DataFrame', size=4122, hash='EzUJIW3AamdtaNxG_Bu_nA', hash_type='md5', visibility=1, key_is_virtual=True, updated_at='2024-05-25 15:24:52 UTC')
Provenance
.created_by = 'anonymous'
.storage = '/home/runner/work/lamindb/lamindb/docs/lamin-intro'
.transform = 'Introduction'
.run = '2024-05-25 15:24:51 UTC'
Labels
.ulabels = 'Candidate marker study'
| uid | name | description | reference | reference_type | run_id | created_by_id | updated_at | |
|---|---|---|---|---|---|---|---|---|
| id | ||||||||
| 1 | Gk5diowj | Candidate marker study | None | None | None | 1 | 1 | 2024-05-25 15:24:53.067259+00:00 |
Queries¶
Write arbitrary relational queries (under-the-hood, LaminDB is SQL & Django).
# get an entity by uid
transform = ln.Transform.get("FPnfDtJz8qbE")
# filter by description
ln.Artifact.filter(description="my RNA-seq").df()
# query all artifacts ingested from a notebook named "Introduction"
artifacts = ln.Artifact.filter(transform__name="Introduction").all()
# query all artifacts ingested from a notebook with "intro" in the name and labeled "Candidate marker study"
artifacts = ln.Artifact.filter(transform__name__icontains="intro", ulabels=candidate_marker_study).all()
Search¶
# search in a registry
ln.Transform.search("intro").df()
# look up records with auto-complete
labels = ln.ULabel.lookup()
Show me a screenshot
Validate & annotate¶
In LaminDB, validation & annotation of categoricals are closely related by mapping categories on registry content.
Let’s validate a DataFrame by passing validation criteria while constructing an Annotate flow object.
Validate¶
# construct an object to validate & annotate a DataFrame
annotate = ln.Annotate.from_df(
df,
# define validation criteria
columns=ln.Feature.name, # map column names
categoricals={df.perturbation.name: ln.ULabel.name}, # map categories
)
# the dataframe doesn't validate because registries don't contain the identifiers
annotate.validate()
Show code cell output
✅ added 1 record with Feature.name for columns: 'perturbation'
❗ 3 non-validated categories are not saved in Feature.name: ['CD14', 'CD4', 'CD8A']!
→ to lookup categories, use lookup().columns
→ to save, run add_new_from_columns
💡 mapping perturbation on ULabel.name
❗ 2 terms are not validated: 'DMSO', 'IFNG'
→ save terms via .add_new_from('perturbation')
False
Update registries¶
# add non-validated identifiers to their mapped registries
annotate.add_new_from_columns()
annotate.add_new_from(df.perturbation.name)
# the registered labels & features that will from now on be used for validation
ln.ULabel.df()
ln.Feature.df()
Show code cell output
✅ added 3 records with Feature.name for columns: 'CD14', 'CD4', 'CD8A'
✅ added 2 records with ULabel.name for perturbation: 'DMSO', 'IFNG'
| uid | name | dtype | unit | description | synonyms | run_id | created_by_id | updated_at | |
|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||
| 4 | GL42DE9mSRxB | CD14 | int | None | None | None | 1 | 1 | 2024-05-25 15:24:53.300746+00:00 |
| 3 | UnmIj4GN91xU | CD4 | int | None | None | None | 1 | 1 | 2024-05-25 15:24:53.300610+00:00 |
| 2 | gWR4P8p9Ekwv | CD8A | int | None | None | None | 1 | 1 | 2024-05-25 15:24:53.300470+00:00 |
| 1 | UPHCdb8aNaS7 | perturbation | cat | None | None | None | 1 | 1 | 2024-05-25 15:24:53.158283+00:00 |
Annotate¶
# given the updated registries, the validation passes
annotate.validate()
# save annotated artifact
artifact = annotate.save_artifact(description="my RNA-seq", version="1")
artifact.describe()
Show code cell output
✅ perturbation is validated against ULabel.name
💡 returning existing artifact with same hash: Artifact(uid='NtOFwmJMkuNYO5rjoQUS', version='1', description='my RNA-seq', suffix='.parquet', accessor='DataFrame', size=4122, hash='EzUJIW3AamdtaNxG_Bu_nA', hash_type='md5', visibility=1, key_is_virtual=True, created_by_id=1, storage_id=1, transform_id=1, run_id=1, updated_at='2024-05-25 15:24:52 UTC')
Artifact(uid='NtOFwmJMkuNYO5rjoQUS', version='1', description='my RNA-seq', suffix='.parquet', accessor='DataFrame', size=4122, hash='EzUJIW3AamdtaNxG_Bu_nA', hash_type='md5', visibility=1, key_is_virtual=True, updated_at='2024-05-25 15:24:53 UTC')
Provenance
.created_by = 'anonymous'
.storage = '/home/runner/work/lamindb/lamindb/docs/lamin-intro'
.transform = 'Introduction'
.run = '2024-05-25 15:24:51 UTC'
Labels
.ulabels = 'Candidate marker study', 'DMSO', 'IFNG'
Features
'perturbation' = 'DMSO', 'IFNG'
Feature sets
'columns' = 'perturbation', 'CD8A', 'CD4', 'CD14'
Query for annotations¶
ulabels = ln.ULabel.lookup()
ln.Artifact.filter(ulabels=ulabels.ifng).one()
Show code cell output
Artifact(uid='NtOFwmJMkuNYO5rjoQUS', version='1', description='my RNA-seq', suffix='.parquet', accessor='DataFrame', size=4122, hash='EzUJIW3AamdtaNxG_Bu_nA', hash_type='md5', visibility=1, key_is_virtual=True, created_by_id=1, storage_id=1, transform_id=1, run_id=1, updated_at='2024-05-25 15:24:53 UTC')
Biological registries¶
The generic Feature and ULabel registries will get you pretty far.
But let’s now look at what you do can with a dedicated biological registry like Gene.
Access public ontologies¶
Every bionty registry is based on configurable public ontologies.
import bionty as bt
cell_types = bt.CellType.public()
cell_types
Show code cell output
PublicOntology
Entity: CellType
Organism: all
Source: cl, 2024-02-13
#terms: 2918
cell_types.search("gamma delta T cell").head(2)
Show code cell output
| ontology_id | definition | synonyms | parents | __ratio__ | |
|---|---|---|---|---|---|
| name | |||||
| gamma-delta T cell | CL:0000798 | A T Cell That Expresses A Gamma-Delta T Cell R... | gammadelta T cell|gamma-delta T-cell|gamma-del... | [CL:0000084] | 100.000000 |
| CD27-negative gamma-delta T cell | CL:0002125 | A Circulating Gamma-Delta T Cell That Expresse... | gammadelta-17 cells | [CL:0000800] | 86.486486 |
Validate & annotate with typed features¶
import anndata as ad
# store the dataset as an AnnData object to distinguish data from metadata
adata = ad.AnnData(df[["CD8A", "CD4", "CD14"]], obs=df[["perturbation"]])
# create an annotation flow for an AnnData object
annotate = ln.Annotate.from_anndata(
adata,
# define validation criteria
var_index=bt.Gene.symbol, # map .var.index onto Gene registry
categoricals={adata.obs.perturbation.name: ln.ULabel.name},
organism="human", # specify the organism for the Gene registry
)
annotate.validate()
# save annotated artifact
artifact = annotate.save_artifact(description="my RNA-seq", version="1")
artifact.describe()
Show code cell output
✅ added 3 records from public with Gene.symbol for var_index: 'CD8A', 'CD4', 'CD14'
✅ var_index is validated against Gene.symbol
✅ perturbation is validated against ULabel.name
💡 path content will be copied to default storage upon `save()` with key `None` ('.lamindb/q0FZOj09RDJdp2gH7qbO.h5ad')
✅ storing artifact 'q0FZOj09RDJdp2gH7qbO' at '/home/runner/work/lamindb/lamindb/docs/lamin-intro/.lamindb/q0FZOj09RDJdp2gH7qbO.h5ad'
💡 parsing feature names of X stored in slot 'var'
✅ 3 terms (100.00%) are validated for symbol
✅ linked: FeatureSet(uid='aNTJxi3V5xrxIyWQQm09', n=3, dtype='int', registry='bionty.Gene', hash='f2UVeHefaZxXFjmUwo9O', created_by_id=1, run_id=1)
💡 parsing feature names of slot 'obs'
✅ 1 term (100.00%) is validated for name
✅ linked: FeatureSet(uid='abFTleHzqedIqwDk9cQ6', n=1, registry='Feature', hash='RpVrTX3vdB_it00v9rhU', created_by_id=1, run_id=1)
✅ saved 2 feature sets for slots: 'var','obs'
Artifact(uid='q0FZOj09RDJdp2gH7qbO', version='1', description='my RNA-seq', suffix='.h5ad', accessor='AnnData', size=19240, hash='ohAeiVMJZOrc3bFTKmankw', hash_type='md5', n_observations=3, visibility=1, key_is_virtual=True, updated_at='2024-05-25 15:24:57 UTC')
Provenance
.created_by = 'anonymous'
.storage = '/home/runner/work/lamindb/lamindb/docs/lamin-intro'
.transform = 'Introduction'
.run = '2024-05-25 15:24:51 UTC'
Labels
.ulabels = 'DMSO', 'IFNG'
Features
'perturbation' = 'DMSO', 'IFNG'
Feature sets
'var' = 'CD8A', 'CD4', 'CD14'
'obs' = 'perturbation'
Query for typed features¶
# get a lookup object for human genes
genes = bt.Gene.filter(organism__name="human").lookup()
# query for all feature sets that contain CD8A
feature_sets = ln.FeatureSet.filter(genes=genes.cd8a).all()
# write the query
ln.Artifact.filter(feature_sets__in=feature_sets).df()
Show code cell output
| uid | version | description | key | suffix | accessor | size | hash | hash_type | n_objects | n_observations | visibility | key_is_virtual | storage_id | transform_id | run_id | created_by_id | updated_at | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||||||
| 2 | q0FZOj09RDJdp2gH7qbO | 1 | my RNA-seq | None | .h5ad | AnnData | 19240 | ohAeiVMJZOrc3bFTKmankw | md5 | None | 3 | 1 | True | 1 | 1 | 1 | 1 | 2024-05-25 15:24:57.419252+00:00 |
Add new records¶
Create a cell type record and add a new cell state.
# create an ontology-coupled cell type record and save it
neuron = bt.CellType.from_public(name="neuron")
neuron.save()
Show code cell output
✅ created 1 CellType record from Bionty matching name: 'neuron'
💡 also saving parents of CellType(uid='3QnZfoBk', name='neuron', ontology_id='CL:0000540', synonyms='nerve cell', description='The Basic Cellular Unit Of Nervous Tissue. Each Neuron Consists Of A Body, An Axon, And Dendrites. Their Purpose Is To Receive, Conduct, And Transmit Impulses In The Nervous System.', created_by_id=1, run_id=1, public_source_id=29, updated_at='2024-05-25 15:24:58 UTC')
✅ created 3 CellType records from Bionty matching ontology_id: 'CL:0000393', 'CL:0002319', 'CL:0000404'
❗ now recursing through parents: this only happens once, but is much slower than bulk saving
💡 you can switch this off via: bt.settings.auto_save_parents = False
💡 also saving parents of CellType(uid='2qSJYeQX', name='electrically responsive cell', ontology_id='CL:0000393', description='A Cell Whose Function Is Determined By Its Response To An Electric Signal.', created_by_id=1, run_id=1, public_source_id=29, updated_at='2024-05-25 15:24:58 UTC')
✅ created 1 CellType record from Bionty matching ontology_id: 'CL:0000211'
💡 also saving parents of CellType(uid='590vrK18', name='electrically active cell', ontology_id='CL:0000211', description='A Cell Whose Function Is Determined By The Generation Or The Reception Of An Electric Signal.', created_by_id=1, run_id=1, public_source_id=29, updated_at='2024-05-25 15:24:59 UTC')
✅ created 1 CellType record from Bionty matching ontology_id: 'CL:0000000'
💡 also saving parents of CellType(uid='7kYbAaTq', name='neural cell', ontology_id='CL:0002319', description='A Cell That Is Part Of The Nervous System.', created_by_id=1, run_id=1, public_source_id=29, updated_at='2024-05-25 15:24:58 UTC')
💡 also saving parents of CellType(uid='5NqNmmSr', name='electrically signaling cell', ontology_id='CL:0000404', description='A Cell That Initiates An Electrical Signal And Passes That Signal To Another Cell.', created_by_id=1, run_id=1, public_source_id=29, updated_at='2024-05-25 15:24:58 UTC')
# create a record to track a new cell state
new_cell_state = bt.CellType(name="my neuron cell state", description="explains X")
new_cell_state.save()
# express that it's a neuron state
new_cell_state.parents.add(neuron)
# view ontological hierarchy
new_cell_state.view_parents(distance=2)
❗ records with similar names exist! did you mean to load one of them?
| uid | name | ontology_id | abbr | synonyms | description | public_source_id | run_id | created_by_id | updated_at | |
|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||
| 1 | 3QnZfoBk | neuron | CL:0000540 | None | nerve cell | The Basic Cellular Unit Of Nervous Tissue. Eac... | 29 | 1 | 1 | 2024-05-25 15:24:58.069701+00:00 |
| 2 | 2qSJYeQX | electrically responsive cell | CL:0000393 | None | None | A Cell Whose Function Is Determined By Its Res... | 29 | 1 | 1 | 2024-05-25 15:24:58.614828+00:00 |
| 3 | 7kYbAaTq | neural cell | CL:0002319 | None | None | A Cell That Is Part Of The Nervous System. | 29 | 1 | 1 | 2024-05-25 15:24:58.614977+00:00 |
| 4 | 5NqNmmSr | electrically signaling cell | CL:0000404 | None | None | A Cell That Initiates An Electrical Signal And... | 29 | 1 | 1 | 2024-05-25 15:24:58.615112+00:00 |
| 5 | 590vrK18 | electrically active cell | CL:0000211 | None | None | A Cell Whose Function Is Determined By The Gen... | 29 | 1 | 1 | 2024-05-25 15:24:59.062755+00:00 |

Scale up data & learning¶
How do you learn from new datasets that extend your previous data history? Leverage Collection.
# a new dataset
df = pd.DataFrame(
{
"CD8A": [2, 3, 3],
"CD4": [3, 4, 5],
"CD38": [4, 2, 3],
"perturbation": ["DMSO", "IFNG", "IFNG"]
},
index=["observation4", "observation5", "observation6"],
)
adata = ad.AnnData(df[["CD8A", "CD4", "CD38"]], obs=df[["perturbation"]])
# validate, annotate and save a new artifact
annotate = ln.Annotate.from_anndata(
adata,
var_index=bt.Gene.symbol,
categoricals={adata.obs.perturbation.name: ln.ULabel.name},
organism="human"
)
annotate.validate()
artifact2 = annotate.save_artifact(description="my RNA-seq dataset 2")
Show code cell output
✅ added 1 record from public with Gene.symbol for var_index: 'CD38'
✅ var_index is validated against Gene.symbol
✅ perturbation is validated against ULabel.name
💡 path content will be copied to default storage upon `save()` with key `None` ('.lamindb/AO8T2VdY2Y6OZdc4zrSl.h5ad')
✅ storing artifact 'AO8T2VdY2Y6OZdc4zrSl' at '/home/runner/work/lamindb/lamindb/docs/lamin-intro/.lamindb/AO8T2VdY2Y6OZdc4zrSl.h5ad'
💡 parsing feature names of X stored in slot 'var'
✅ 3 terms (100.00%) are validated for symbol
✅ linked: FeatureSet(uid='gslsAA4ufbfYwLvSD2Cj', n=3, dtype='int', registry='bionty.Gene', hash='QW2rHuIo5-eGNZbRxHMD', created_by_id=1, run_id=1)
💡 parsing feature names of slot 'obs'
✅ 1 term (100.00%) is validated for name
✅ linked: FeatureSet(uid='abFTleHzqedIqwDk9cQ6', n=1, registry='Feature', hash='RpVrTX3vdB_it00v9rhU', created_by_id=1, run_id=1)
✅ saved 1 feature set for slot: 'var'
Collections of artifacts¶
Create a collection using Collection.
collection = ln.Collection([artifact, artifact2], name="my RNA-seq collection", version="1")
collection.save()
collection.describe()
collection.view_lineage()
Show code cell output
✅ saved 1 feature set for slot: 'var'
Collection(uid='AeopTrA50IvDlXjQk2bM', version='1', name='my RNA-seq collection', hash='5g0aLY_lBSTkIYYUTycd', visibility=1, updated_at='2024-05-25 15:25:02 UTC')
Provenance
.created_by = 'anonymous'
.transform = 'Introduction'
.run = '2024-05-25 15:24:51 UTC'

# if it's small enough, you can load the entire collection into memory as if it was one
collection.load()
# typically, it's too big, hence, iterate over its artifacts
collection.artifacts.all()
# or look at a DataFrame listing the artifacts
collection.artifacts.df()
Show code cell output
| uid | version | description | key | suffix | accessor | size | hash | hash_type | n_objects | n_observations | visibility | key_is_virtual | storage_id | transform_id | run_id | created_by_id | updated_at | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||||||
| 2 | q0FZOj09RDJdp2gH7qbO | 1 | my RNA-seq | None | .h5ad | AnnData | 19240 | ohAeiVMJZOrc3bFTKmankw | md5 | None | 3 | 1 | True | 1 | 1 | 1 | 1 | 2024-05-25 15:24:57.419252+00:00 |
| 3 | AO8T2VdY2Y6OZdc4zrSl | None | my RNA-seq dataset 2 | None | .h5ad | AnnData | 19240 | L37UPl4IUH20HkIRzvlRMw | md5 | None | 3 | 1 | True | 1 | 1 | 1 | 1 | 2024-05-25 15:25:02.951755+00:00 |
Data loaders¶
# to train models, batch iterate through the collection as if it was one array
from torch.utils.data import DataLoader, WeightedRandomSampler
dataset = collection.mapped(obs_keys=["perturbation"])
sampler = WeightedRandomSampler(
weights=dataset.get_label_weights("perturbation"), num_samples=len(dataset)
)
data_loader = DataLoader(dataset, batch_size=2, sampler=sampler)
for batch in data_loader:
pass
Read this blog post for more on training models on sharded datasets.
Data lineage¶
Save notebooks & scripts¶
If you call finish(), you save the run report, source code, and compute environment to your default storage location.
ln.finish()
See an example for this introductory notebook here.
Show me a screenshot
If you want to cache a notebook or script, call:
lamin get https://lamin.ai/laminlabs/lamindata/transform/FPnfDtJz8qbE5zKv
Data lineage across entire projects¶
View the sequence of data transformations (Transform) in a project (from a use case, based on Schmidt et al., 2022):
transform.view_parents()

Or, the generating flow of an artifact:
artifact.view_lineage()

Both figures are based on mere calls to ln.track() in notebooks, pipelines & app.
Distributed databases¶
Easily create & access databases¶
LaminDB is a distributed system like git. Similar to cloning a repository, collaborators can connect to your instance via:
ln.connect("account-handle/instance-name")
Or you load an instance on the command line for auto-connecting in a Python session:
lamin load "account-handle/instance-name"
Or you create your new instance:
lamin init --storage ./my-data-folder
Custom schemas and plugins¶
LaminDB can be customized & extended with schema & app plugins building on the Django ecosystem. Examples are:
bionty: Registries for basic biological entities, coupled to public ontologies.
wetlab: Exemplary custom schema to manage samples, treatments, etc.
If you’d like to create your own schema or app:
Create a git repository with registries similar to wetlab
Create & deploy migrations via
lamin migrate createandlamin migrate deploy
It’s fastest if we do this for you based on our templates within an enterprise plan.
Design¶
Why?¶

The complexity of modern R&D data often blocks realizing the scientific progress it promises: see this blog post.
More basically: The pydata family of objects is at the heart of most data science, ML & comp bio workflows: DataFrame, AnnData, pytorch.DataLoader, zarr.Array, pyarrow.Table, xarray.Collection, etc. We couldn’t find a tool to link these objects to context so that they could be analyzed in context:
provenance: data sources, data transformations, models, users
domain knowledge & experimental metadata: the features & labels derived from domain entities
Assumptions¶
Schema & API¶
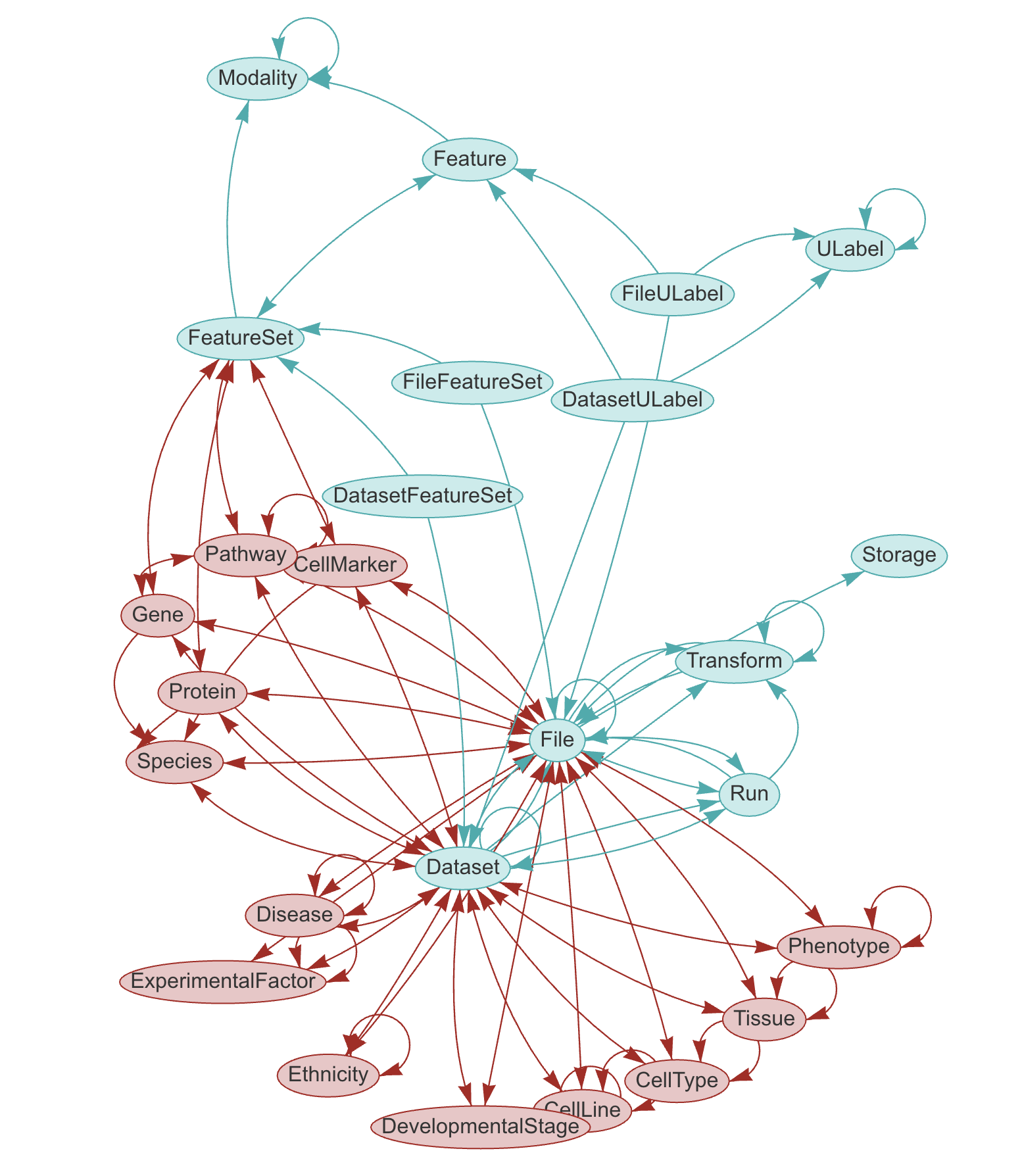
LaminDB provides a SQL schema for common entities: Artifact, Collection, Transform, Feature, ULabel etc. - see the API reference or the source code.
The core schema is extendable through plugins (see blue vs. red entities in graphic), e.g., with basic biological (Gene, Protein, CellLine, etc.) & operational entities (Biosample, Techsample, Treatment, etc.).
What is the schema language?
Data models are defined in Python using the Django ORM. Django translates them to SQL tables. Django is one of the most-used & highly-starred projects on GitHub (~1M dependents, ~73k stars) and has been robustly maintained for 15 years.
On top of the schema, LaminDB is a Python API that abstracts over storage & database access, data transformations, and (biological) ontologies.
Repositories¶
LaminDB and its plug-ins consist in open-source Python libraries & publicly hosted metadata assets:
lamindb: Core API, which builds on the core schema.
bionty: Registries for basic biological entities, coupled to public ontologies.
wetlab: An (exemplary) wetlab schema.
guides: Guides.
usecases: Use cases.
LaminHub is not open-sourced.
Influences¶
LaminDB was influenced by many other projects, see Influences.